

My recent talk in Copenhagen
My recent talk in Copenhagen
Failure of drug regulation; declining standards and institutional corruption


Recently, I presented a talk in Copenhagen, Denmark, titled, “Failure of Drug Regulation: Declining standards and institutional corruption”
It was hosted by the Centre for Evidence-Based Medicine (CEBM), Oxford and the Institute for Scientific Freedom, Denmark.
SLIDES below:
“Safe and effective” is a phrase adopted by drug regulators to assure the public that the pills we reach for in our medicine cabinets, or the vaccines injected into our arms, have proven benefits that outweigh the risks. And we assume this is based on a rigorous, independent assessment of the trial data. But the information I will present today, demonstrates that drug regulators have fallen victim to institutional corruption and have often violated their sworn oath to protect public health.

My recent investigation for The BMJ analysed six major international drug regulators, comparing aspects such as conflicts of interest, proportion of industry funding, transparency of data and drug approval pathways.

A summary of what I found is in Table 1. Notably, you will see in the top row, that all the major drug regulators are largely funded by the very industries they are sworn to regulate, with the Australian TGA receiving 96% of its funds from the drug industry. In this talk, however, I will focus on the US FDA, because it is the most well-funded, well-resourced regulator in the world, and low to middle income countries look to the FDA for regulatory decisions.
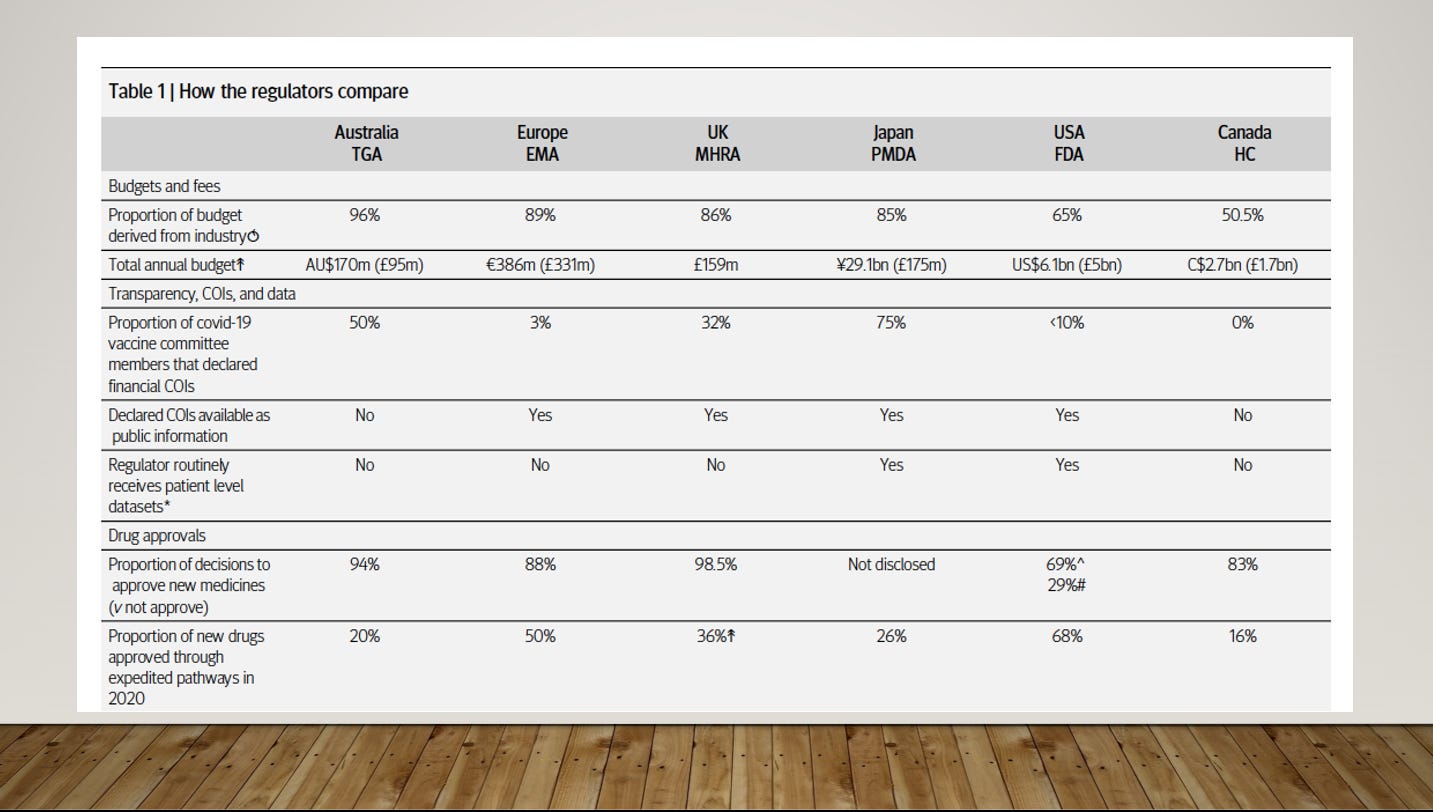
Following the HIV/AIDS crisis in the 80s there was concern, especially from advocacy groups, that it was taking too long to get drugs to market. US Congress passed the 1992 Prescription Drug User Fee Act allowing the FDA to accept industry funding. Fees increased 30-fold from $29m in 1993 to $884m in 2016. Now, 65% of funding for drug evaluation comes from industry fees. The FDA hired more staff so that it could speed up approvals. It worked. In 1988, only 4% of drugs introduced onto the market were approved first by the FDA, but that rose to 66% by 1998 after its funding structure changed.

Now, 68% of drugs approved by the FDA are via expedited pathways. Advocacy groups are especially pleased because it means that people gain faster access to medicines. The downside however, is that expedited approvals require a lower burden of proof.
One example is how the FDA accepts changes in a ‘surrogate marker’ rather than a ‘clinical outcome.’ The drug Aducanabab for Alzheimers was approved on the basis of lowered beta-amyloid proteins, without any meaningful improvement to the patients symptoms.
It means drug companies require fewer, smaller, less robust trials to get their drugs into the marketplace.
The FDA also allows drugs onto the market before efficacy has been proven, on the condition that drug companies perform "confirmatory trials”. However, these trials are often not carried out, or they take years to show the drug doesn’t work, all the while, the drug is being sold on the market under the pretense of ‘safe and effective.’
Consequently, drugs approved via expedited pathways, are more likely to be withdrawn for safety reasons or have black box warnings.

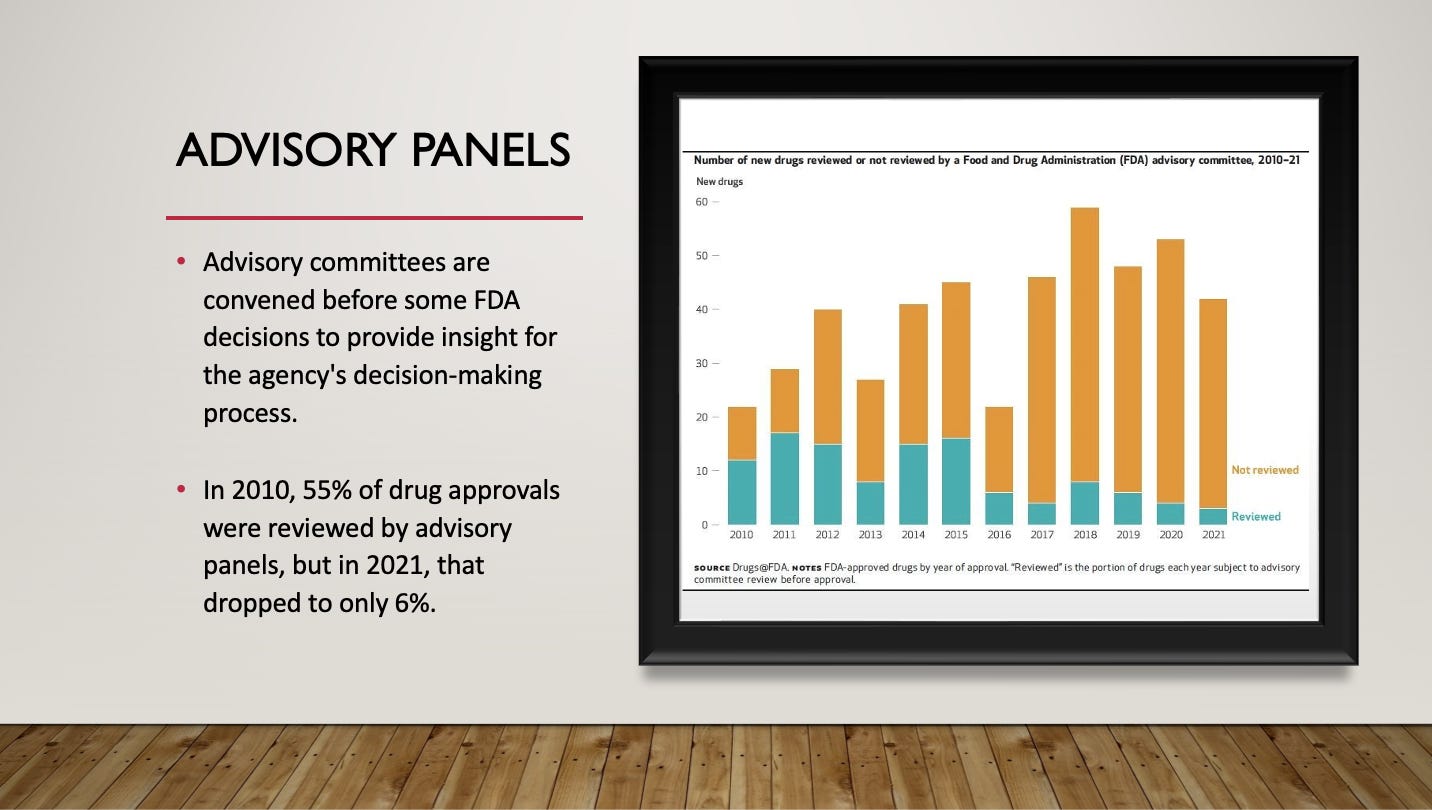
During the drug approval process, the FDA convenes advisory panels to provide insight and credibility to the decision-making process. But a recent analysis shows that the FDA is seeking this independent advice, less frequently. In 2010, 55% of drug approvals were reviewed by advisory panels, but in 2021, that dropped to just 6%.
The FDA conducts random inspections of clinical trial sites to ensures the integrity of data from those trials. If there are any violations or objectional practices, the FDA writes up an inspection report. The reports are not proactively shared with the public, and often, details are only obtained by filing an FOIA request. FOIAs can take several months to be processed, and reports end up highly redacted, and of limited value (as shown on the slide).
In 2007, the Office of Inspector General determined that the FDA only conducted inspections at 1% of trial sites and there was no database for how many sites were operating across the US and abroad. The FDA says it doesn’t know how many trial sites are operating and that keeping track of them all is ‘too resource intensive.’

During the pandemic, things went from bad to worse for the FDA. While vaccine manufacturers ramped up their operations, the restrictions meant that the FDA’s onsite inspections of trial sites ground to a halt for a period of time. Regulatory documents show that the FDA only inspected 9 in 153 trial sites for Pfizer and 1 in 99 sties for Moderna [CORRECTION: only 1 site was inspected prior to full approval, but 9 were inspected for EUA, making a total of 10 sites]

Notably, a whistle-blower named Brook Jackson, lodged a complaint with the FDA about falsified data at three of Pfizer’s mRNA trial sites, based in Texas. She was the regional director at the three trial sites and witnessed falsification of data, the unblinding of patients, and inadequately trained vaccinators who failed to follow up on adverse events. Alarmingly, the FDA never conducted an onsite inspection of those sites after Jackson’s complaint.

It is clear that the FDA’s evidentiary standards have declined over the decades. The agency relies heavily on drug industry money, which has led to faster drug approvals but the drugs are more unsafe. The FDA is relying less on its external advisory committees when approving drugs and its no secret that the agency has endured enormous pressure from the White house during the pandemic. Not just from the Trump administration, but from the Biden Administration too. Top vaccine officials, Marion Gruber and Philip Krause resigned in October last year, after Biden pushed a widespread booster roll out in spite of the evidence. A Government accountability report this year revealed that staffers at the agency were under enormous political pressure, which they say, resulted in the “suppression of scientific findings” regarding covid-19.

Reform is tricky - some suggest incremental improvements in transparency of data and conflicts of interest. Others suggest there needs to be an independent body assessing the FDA’s drug approval decisions. It has also been suggested that the FDA needs a major overhaul, that the agency needs to ‘clean house’ and rid itself of bureaucrats who are simply biding their time until retirement. There are no clear answers but most of the people I interviewed over the last few months agree that the agency is “broken”.

If you have thoughts on how to improve the drug regulatory system, then please leave comments below.
In the past we made a decision to give the pharmaceutical companies a monopoly via patents on drugs they developed. We'd decided the trade-off between paying excess in prices was worth it to get the new drugs developed, and to let the industry that benefited pay the cost of developing new drugs, including all the ones that did not pan out. However, the industry has changed a lot over time. Scientists are, relatively speaking, low cost. The computer simulations which used to need extremely expensive computer time on mainframes can now be done on consumer-level machines. And new drug candidates are only rarely the result of some new imaginative insight. More often they are a result of a brute-force technique of trying chemical compounds which are slightly different than the ones already in use. The name of the game here is not to produce a drug which is better than existing ones, though it is nice when that happens, but one that can be patented and sold as a replacement for something that either your competitor has and is making money with, or that you have, but whose patent is due to run out.
Thus for the creative end of drug discovery, the pharmaceutical companies do not need the monopoloy windfall profits. The expensive part of the operation is the trials. But here we have a fundamental misalignment between the goals of the industry and the public weal. The trials are either run by drug companies, or outsourced to testing companies who work for drug companies. And their business is to get the drug approved, and promise to run the trials and only present the data that shows the drugs in a good light and so on and so forth. 'Can we fool the regulators into accepting this?' used to be the name of the game. 'Can we bribe the regulators into accepting this?' is the newer game, and it works.
So. We need to strip the monopoly protection from pharmaceuticals. We need to have the testing done by a new, taxpayer funded organisations (who are allowed to outsource this). We can tax the drugs to afford the trials. We need to work very hard to keep regulatory capture from happening in the new organisations, and to notice it when corruption starts up again, as always happens. This will entail as robust a set of transparency laws as possible, so we can get rid of the secrecy around all of this. Patenting was _supposed_ to give us full disclosure, as that was the deal, you give us full disclosure and we give you a patent. I do not know if this ever worked in the past, but it is not working now where we need FOI requests to find out what ought to be public knowledge. Laws prohibiting the revolving door between regulators and industry and preventing regulators from owning stock in the companies they regulate will be necessary. Over time the problem will lessen as drug companies just aren't that rich any more.
Take away the monopoly protection and the secrecy. We tried the experiment of building the industry this way, and now we know that it does not work in the public interest. Time to do it another way.
You're doing very important work! Love it!